2016年度生物測定学応用実験
分子系統樹の作成
東京大学大学院農学生命科学研究科 大森宏
http://lbm.ab.a.u-tokyo.ac.jp/~omori/phylogeny/txt/phylo.html
2016年11月24日
1. はじめに
今回の実験では,データベースからインターネットにより
DNA シーケンスデータを取得し,そのデータから遺伝子系統樹を
作成する実験を行う.手元になにも情報がない状態から、
どのように情報を得るかを学び,合わせて,分子系統樹が実際のデータから
どのように作成されるかを見ることを目的としている.
2. シーケンスデータの取得
サルのリボヌクレアーゼスーパーファミリー
RNA 分解酵素(リボヌクレアーゼ (RNase))ファミリーである ECP (eosinophil cationic protein)
と EDN (eosinophil-derived neurotoxin) は,エオシン好性白血球 (eosinophil leukocyte) という
顆粒白血球に局在している.EDN の生理機能はよくわかっていないが,リボヌクレアーゼと
しての活性は高く,レトロウィルスのゲノム RNA
を破壊することにより一種の抗ウィルス剤として働いているのではないかという報告が
なされている.一方,ECP はリボヌクレアーゼの活性は低いが,病原性バクテリアや寄生虫
の細胞膜に穴をあけて殺す機能があり,これらの病原体に対する強力な防御を行っている.
今回の実験では,ヒトとサルの EDN と ECP のシーケンスデータをゲノムデータベース
からインターネットで取得
して,分子系統樹を作成する.用いる生物種は,
- human Homo sapiens
- chimpanzee Pan troglodytes
- gorilla Gorilla gorilla
- orangutan Pongo pygmaeus
- macaque Macaca fascicularis,ニホンザル,タイワンザルなど
- tamarin Saguinus oedipus,シシザル
である.ECP は1から5までの生物種にあり,EDN は上に挙げたすべての生物種にある.
ヒト DNA データの取得
以下のようにして,シーケンスデータをインターネットを
用いてデータベース
から取得する.一つのサイトに集中すると迷惑なので,各自で以下のサイトを適宜選ぶ.
ヒトの場合について述べる.塩基配列には偽遺伝子があったり,イントロンも含まれている
ので,基本的にはなるべく短い配列がよいと考えられる.
1.ゲノムネット(京都大学化学研究所バイオインフォマティックスセンター)
- 京都大学ゲノムネット(http://www.genome.jp/ja/) に行く.
-
`Search' ボックスを「塩基配列」にする.
- `for' ボックスの中に `eosinophil human' と書く.
- `eosinophil cationic protein' で検索してもよい.
- `Go' をクリックする.
- 検索された中から良さそうなものを見に行く.([GenBank],[EMBL],[DDBJ] なんでも良い)
- 左上の`FASTA' を見に行く.(GenBank の場合)
- もしくは,`ORIGIN' を見に行く.
- 塩基配列をコピーして,テキストエディターに
張り付ける.
- humanEDN.txt' などという名前をつけて適当な場所に保存する.
2.DDBJ(DNA Data Bank of Japan )
-
DDBJ(http://www.ddbj.nig.ac.jp/)に行く.
- 真ん中辺の`検索・解析' をみる。
- `データベース検索' の `ASRS' (高速なキーワード検索 )をクリック.
- QuickSearch に `eosinophil' と `human' を入れて `Search' をクリック.
- 検索された中から良さそうなものを見に行く.
- `ORIGIN' を見に行く.
- 塩基配列をコピーして,メモ帳などのテキストエディターに
張り付ける.
- humanEDN.txt' などという名前をつけて適当な場所に保存する.
3.NCBI (National Center for Biotechnology Information)
-
NCBI (http://www.ncbi.nlm.nih.gov/) に行く.
- `DNA & RNA' の `GenBank' に行く.
他の種のシークエンスデータの取得
ヒトの遺伝子配列が入手できたので,他の種の配列データをを取得する.
ヒトの配列を取得したのと同様の方法(`eosinophil gorilla' で検索)で取得できる.
また,相同配列検索 `BLAST' を用いてもよい.
`BLAST' の使い方(ゲノムネットの場合)
- 下の方の「配列解析」を見る.
- `BLAST' をクリック.
- `BLASTN' をチェックする.
- Human の配列を `Sequence data' ボックスにコピーして `compute' ボタンを押し実行させる.
- 他の生物種の配列を捜す.
3. 系統樹作成
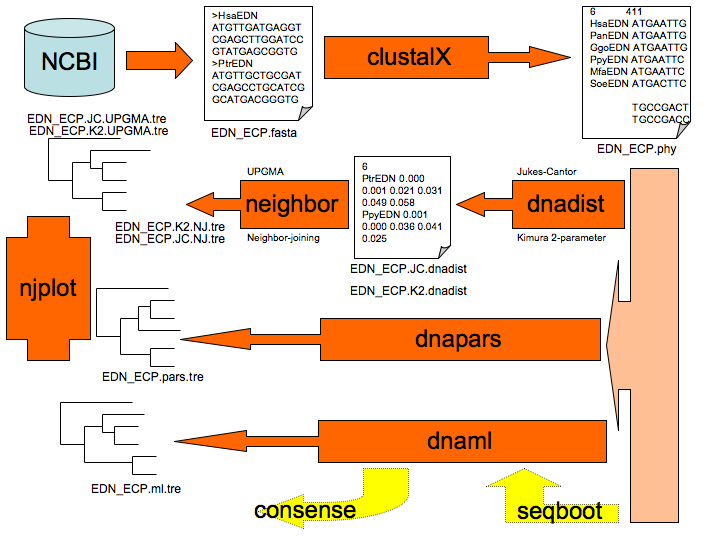
系統樹作のおおまかな流れ
(作図:生物測定研卒業生林氏)
ソフトウェア(Windows)
系統樹を作成するためのフリーのソフトウェアはネット上にある.
`google' などの検索サイトを利用すれば手軽にダウンロードできる.
今回の実験のためのソフトは,
に用意してあるので,ここからダウンロードする.
注)プログラムはダウンロードフォルダにダウンロードされる.
ソフトウェアとデータは同じフォルダ(デスクトップなど)に置く.
ソフトウェア(MAC OS X用)(生物測定研卒業生林氏指南)
ダウンロードされたファイルはデスクトップまたはダウンロードフォルダに保存されます。
ダウンロードされたファイルをダブルクリックするとマウントまたは解凍されます。
中に入っているファイルのうち、.appで終わる次のファイルをデスクトップにコピーしましょう。

テキストエディット.appをDockに表示させておくと便利です.
ソフトウェアとデータは同じフォルダ(デスクトップなど)に置く.
アラインメント clustalX
取得した相同領域のシーケンスの長さが種により異なるときは,アラインメントを
行って長さを揃える必要がある.
一般に3種以上の種を同時に扱うときはマルティプルアラインメトをしなければ
ならない.これは,clustalX というソフトウエアを用いて行う.
- 新規のメモ帳を開き,取得した生物種の配列データをコピーして順に貼り付けて行く.
- `>' を書き,その後に10文字以内で配列名を記入する.
- 系統樹をつくる
配列データ
ができたら,適当な名前で保存する.
- `clustalX' を用いてアラインメントを行う.結果は、`PHYLIP' 形式で
保存する.
- `clustalX' をクリックして開く.
- `File' メニューの `Load sequences' で配列データを入力する.
- `Alignment' メニューの `Output Format Options' をクリックし、`PHYLIP format' にチェックを入れる.
- `Alignment' メニューの `Do Complete Alignment' を選ぶ.
注意)リッチテキスト(.rtf)だと入力できません.マックのテキストエディターのフォーマット
の所を標準テキストにしておくこと.
アライメントをかける配列データ例
アラインメントしたデータを PHYLIP format にした例
このデータ形式にすれば系統樹作成ソフト PHYLIP に入力できる.
進化距離の推定 DNADIST
`DNADIST' を用いて,進化距離 d を以下のようにして計算する.
距離の計算オプションとして,
Junkes-Cantor 法,Kimura's 2 parameter 法の2通りを試す.
- `DNADIST' をクリックする.
- `filename' に先程作成したシーケンスデータを指定する.
- オプションを入力する.
- `y' を打ち込んで実行させる.
- 結果はフォルダ内の `outfile' にできる.
- `outfile' をメモ帳で見て確認し,問題がなければ適当な名前に変えておく.
系統樹作成ソフト phylip
今回の実験で用いるソフトは以下のものである.
- NEIGHBOR
距離行列から系統樹を作成する.UPGMA と Neibor Joining Method (最小進化) が選べる.
- DNAPARS
塩基配列から,最大節約法により系統樹を作成する.
- DNAML
塩基配列から,最尤法(最大確率)により系統樹を作成する.
- NJplot/TreeView
生成した `tree' ファイルから系統樹をきれいに表示する.
PYLIP が生成した "treefile" の名前を変え、拡張子を ".tre" にして読み込ませる.
DNA データの取得と系統樹作成のビデオ
当研究室卒業生林氏のご尽力により,系統樹作成のビデオが YouTube にアップロード
されています.参考にどうぞ.
http://jp.youtube.com/watch?v=vfNFkn5cuIM
動画画面右下の▲にマウスを載せると表示されるうちの下側のボタン(キャプ
ション機能をオンにする)をクリックすると字幕の説明が表示されます.
そのままだと画質が粗いですが、同じく右下の「高画質で表示する」をクリック
すると、オリジナルと同等の高画質動画で再生されます.
- 課題1: (提出不要)生物種1-6のEDN遺伝子に対し,
- 適当な置換モデルを選び,
UPGMA による系統樹をつくれ.
- 適当な置換モデルを選び,
Neibor Joining Method による系統樹をつくれ.
- 最大節約法で系統樹をつくれ.
- 最尤法で系統樹をつくれ.
課題2. 系統樹による EDN と ECP の関係の考察(提出)
RNA 分解酵素(リボヌクレアーゼ (RNase))ファミリーでのうち EDN (eosinophil-derived neurotoxin)
の配列をサルで取得し,系統樹を作成した.一方,RNA 分解酵素ファミリーには ECP (eosinophil
cationic protein) もある.EDN は取り上げた 6 種のサルすべてにあるが、
ECPはシシザルには無く 5 種のサルにある.
ECP は EDN からの遺伝子重複により形成されたと考えられている.
そうすると,遺伝子重複後 EDN から ECP に変化した遺伝子系統は配列が大きく変化したと考えられるので、進化
距離が大きくなっていることが予想される.これを確かめるには,EDN と ECP を混ぜて系統樹をつくればよい.
- 系統樹の作成手法を UPGMA,NJ,最尤法の中から1つ選び.EDN と ECP を混ぜて系統樹を作成せよ.
EDN と ECP とにどのような関係があるか,系統樹を見て論ぜよ.ただし,「遺伝子重複」と「進化距離」の2文字
は必ず使用すること.
課題3. ブートストラップ法による分岐の信頼性の評価(発展)
分子系統樹を作成したときに,その分岐パターンがどの程度信頼性があるか調べる統計的手法.
いま,系統樹を n サイトで構成したとき,この n サイトから繰り返しを許して n サイトをリサンプル
して系統樹を作成する.リサンプルされた系統樹で同一の分岐パターンが得られればその分岐パターン
はそれだけ信頼性がおけることになる.
サル族の EDN と ECP の系統樹にブートストラップ法を適用し,分岐の信頼性を調べる.
参考(当研究室卒業生林氏作成ビデオ)
http://www.youtube.com/watch?v=-YXjOFQ0dds
- ブートストラップ配列データの生成:`seqboot' を起動.
phylip 形式の配列データセットから、サイトのリサンプリングにより、100の配列データセット
を生成する.
- UPGMA, NJ 系統樹:`dnadist' を起動.100の配列データセットそれぞれに対して、距離行列を生成する.
オプションは `M',データセット数は,`100',適当な奇数の値を入れる.
データかウエイトは,`D',データセット数は,`100' を入力する.できあがった距離行列セットファイルを
適当な名前に変え、`neghibor' の入力データとする.オプションは `M',
- 最尤法系統樹:`dnaml' を起動.100の配列データセットそれぞれに対して、最尤法による系統樹を生成する.
オプションは `M',
データかウエイトは,`D',データセット数は,`100',適当な奇数の値を入れ,jumble には 1 を指定する.
- 最頻出枝の出現回数: `consense' を起動.
先ほどの `neghibor' か `dnaml' で得られた tree ファイルを入力すると,枝ごとのブートストラップ
回数が出力される.
- 最初の配列データセットから得られたUPGMA, NJ 系統樹もしくは最尤系統樹の分岐のところに,[ ] をつけ,その中にブートストラップ確率を記入し,
NJtree で見る.
Copyright (C) 2004, Hiroshi Omori. 最終更新:2016年 1月 5日